

Ce nouveau champ d’études élabore des arbres généalogiques à partir des traces relevées dans le génome du virus et d’algorithmes
Le grand public est habitué, grâce aux faits divers ou aux séries télé, à ce que l’ADN aide à confondre les coupables. Mais avec l’épidémie de Covid-19, un autre genre de police génétique est à la manœuvre. S’il ne fait aucun doute que le virus SARS-CoV-2 est bien le responsable de près de 165 000 morts, son patrimoine génétique est en train de révéler aux spécialistes quand il a infecté l’homme, d’où il vient, à quelle vitesse il se répand, combien de gens il a touchés…
Ces nouveaux policiers sont des phylodynamiciens, les représentants d’une discipline qui n’a pas vingt ans et qui montre tout son potentiel avec la pandémie en cours. La découverte du probable passage d’une chauve-souris à l’humain ? C’est la phylodynamique. L’origine d’une contamination dès novembre 2019 en Chine ? C’est encore elle. Les signes de ralentissement de l’épidémie dans certains pays ? Toujours elle. Les origines multiples de l’épidémie en France ? Encore et toujours elle…
« L’idée de la phylodynamique est que la manière dont les virus se propagent laisse des traces dans leur génome », précise Samuel Alizon, chercheur CNRS de l’équipe Evolution théorique et expérimentale du laboratoire Maladies infectieuses et vecteurs : écologie, génétique, évolution et contrôle, à Montpellier. Ces traces sont si infimes qu’il convient de les examiner avec précaution pour les faire « parler », sous peine de se tromper lourdement. Il s’agit de tout petits changements dans l’enchaînement des quelque 30 000 « lettres » qui constituent le génome de ce virus. Une lettre seulement de différence entre deux génomes est déjà une information précieuse.
Une discipline à manier avec précaution
Dès le 20 janvier, une des vedettes du domaine, Trevor Bedford, du centre anticancer Fred Hutchinson, à Seattle (Etas-Unis), acquiert, comme il l’écrit sur son blog, la certitude que ce virus qu’il surveille depuis début janvier est transmissible à l’homme. Une propriété fondamentale qui conditionne la gravité de la maladie à l’échelle planétaire. La Chine ne décrétera une quarantaine dans son premier foyer que le 29 janvier.
L’intuition de ce scientifique vient de l’analyse des génomes viraux qui arrivent à partir du 10 janvier de Chine. Ils sont trop semblables pour croire que les malades auraient été contaminés par des animaux. En effet, le virus étant installé depuis longtemps dans ce réservoir, il devrait exister sous des formes assez variées. Or cette diversité est absente des génomes viraux prélevés sur les premiers malades. Sauf à imaginer qu’un même animal ait contaminé autant d’hommes en des endroits distincts, il fallait se rendre à la terrible évidence que le coronavirus avait trouvé un nouvel hôte et qu’il était devenu transmissible.
Le même spécialiste va très vite mener une seconde enquête pour couper court cette fois à une rumeur. Le 31 janvier, une équipe indienne assure que le génome viral aurait des points communs avec celui du VIH, sous-entendant une manipulation génétique artificielle. Trevor Bedford, dès le lendemain de cette « parution » (l’article a été mis en ligne uniquement sur un site de spécialistes, sans évaluation par une revue scientifique), démonte sur Twitter l’hypothèse. Le genre de variations constatées existe aussi naturellement chez un coronavirus de chauve-souris. Et les Indiens auraient mal comparé les séquences entre elles, prenant un artefact dénué de sens pour une similitude riche d’informations. L’épisode montre que la phylodynamique est à manier avec précaution.
Grave erreur
Le 10 avril, nouvel exemple des subtilités de la technique. Une autre vedette du domaine, Andrew Rambaut, de l’université d’Edimbourg, cloue au pilori des collègues américains pensant avoir découvert trois variants différents dans les diverses souches de virus, comme ils le prétendent dans une grande revue, PNAS. « Ce qui m’énerve le plus est que ces auteurs ont pris quelques données dans une base, les ont mises dans un logiciel facile à utiliser, ont fait des hypothèses inappropriées et publié ce qu’ils ont trouvé. »
Le spécialiste note aussi une grave erreur sur la comparaison avec le coronavirus de la chauve-souris. « Ça me rend un peu triste d’être membre de cette communauté scientifique », déplore sur Twitter un autre spécialiste, François Balloux, professeur de bio-informatique à l’University College de Londres.
L’art phylodynamique est donc subtil. Depuis le début de l’épidémie de Covid-19, l’écart entre les nouveaux génomes et le premier est de moins de vingt lettres de différence sur 30 000 environ. Soit d’un ordre de grandeur analogue au taux d’erreur des techniques de séquençage, d’environ une lettre erronée sur 10 000 lues. A l’inverse, cet écart est de plus de 1 000 lettres entre le probable réservoir du coronavirus, la chauve-souris rhinolophe du Yunnan, et le premier humain touché.
Calculer la vitesse des mutations
« Une centaine de séquences génétiques contient autant d’informations que celles recueillies avec l’ensemble des cas dépistés », assure Samuel Alizon pour défendre l’intérêt de sa discipline dans la compréhension des épidémies. Aux premières observations et controverses ont donc très vite succédé de nouvelles informations, au fur et à mesure qu’arrivaient les séquences du monde entier. Le site Gisaid, de dépôt de ces séquences, en comptait 9 300 le 16 avril ! « Pour un de mes articles dans Science en 2009 sur la grippe A de type H1N1, nous en avions… 11 !, se souvient François Balloux, alors à l’Imperial College. En ce moment, mon équipe traite près de 1 000 séquences par jour. »
Une des premières informations que ces chercheurs extraient concerne la vitesse des mutations, c’est-à-dire le nombre de changements de lettre par an. Plus il y aura de génomes, meilleur sera le calcul, puisqu’il s’agit de faire des statistiques sur l’évolution des écarts entre la séquence d’origine et les nouvelles. On approcherait un taux de changement annuel de 0,08 %. C’est légèrement moins que la grippe ou que le VIH, mais beaucoup plus que pour le génome humain. Le détail est important, car il donne l’horloge moléculaire du virus et permet notamment de remonter le temps.
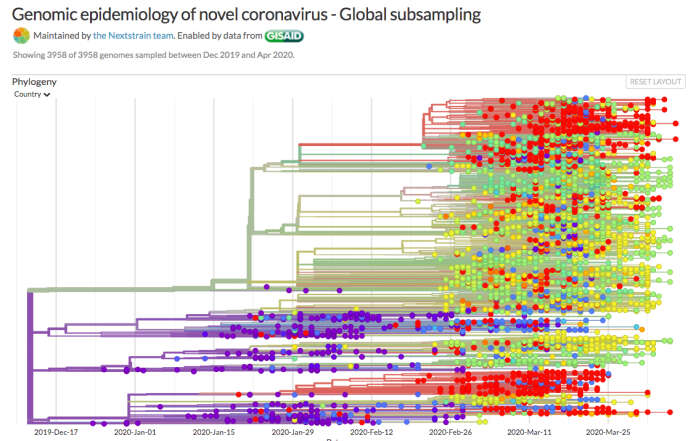
Connaissant le tempo et les dernières séquences, on peut remonter à l’origine temporelle. Andrew Rambaut a ainsi calculé, à partir de 176 génomes, une arrivée probable du virus chez l’homme entre fin août et début décembre 2019, avec une plus forte probabilité pour novembre, bien avant l’identification du premier cas. Sa collègue Tanja Stadler, de l’Ecole polytechnique fédérale de Zurich, qui a étudié 128 génomes, tombe sur la même date, entre novembre et mi-décembre.
Ces mutations, ou substitutions, sont aussi étudiées d’une autre manière. Leur rythme est une chose, leur localisation sur le long brin d’ARN (acide ribonucléique) du virus en est une autre. Chaque partie de cette séquence code en effet pour la fabrication des protéines nécessaires au cycle du virus : accrochage sur la cible, pénétration, détournement du matériel de l’hôte, réplication, prolifération de nouveaux virus et expulsion vers d’autres cellules.
Moins d’une trentaine de protéines sont ainsi déjà identifiées, mais leur rôle n’est pas toujours défini. Certaines mutations sont neutres, c’est-à-dire qu’elles n’influencent pas le comportement du virus. D’autres peuvent en changer la nature et donc sa dangerosité ou sa contagiosité… « Pour l’instant, rien de tel n’a été signalé. On a juste quatre séquences qui montrent des changements sur une protéine-clé, mais rien de confirmé », précise François Balloux. Lui s’intéresse aux parties stables du virus, car ce seront autant de cibles pertinentes pour un éventuel remède. S’il visait des portions trop changeantes, le vaccin ou le médicament perdrait vite de son efficacité.
Des algorithmes pour combler les trous
Mais la vraie force de la phylodynamique est de mélanger ces informations à la fois de temps et « d’espace ». Une autre discipline l’aide dans cette tâche : les mathématiques. Comme des généalogistes, les experts veulent savoir quels sont les « parents probables » d’une séquence, c’est-à-dire les souches à partir desquelles elle a évolué. Ils cherchent donc à placer sur un plan à deux dimensions chacun des génomes viraux d’individus prélevés et voir en quoi ils partagent les mêmes mutations et quels liens les relient.
Apparaît alors un arbre, avec des « feuilles », qui sont les génomes séquencés, des rameaux, puis des branches plus ou moins regroupées, plus ou moins longues, selon qu’elles correspondent à un petit ou grand nombre de mutations. Mais comme on ne peut disposer de toutes les séquences de tous les individus, cet « arbre » parfait est hors de portée. Les trous doivent être comblés par les mathématiques.
Des algorithmes essaient d’inférer les liens entre feuilles en proposant l’arbre généalogique le plus probable correspondant aux données recueillies. Ce qui veut dire qu’à chaque modèle mathématique distinct, un arbre aux embranchements légèrement différents peut être obtenu. Cette reconstruction, proposée en 1981 par Joseph Felsenstein, a véritablement ouvert le domaine… vingt ans plus tard ! Non seulement il fallait attendre d’avoir assez de séquences, mais en plus les calculs demandaient une puissance informatique inexistante à l’époque.
Aujourd’hui, tout est réuni. Les spécialistes font pousser des arbres à foison. Même le grand public peut saisir toute la puissance de ces nouvelles analyses grâce au site Nextstrain, qui collecte les génomes de Gisaid, puis les traite grâce à divers algorithmes pour réaliser ces arbres, joliment présentés.
Il saute ainsi aux yeux, ou presque, que les contaminations des Etats-Unis ont eu plusieurs origines. Tout comme en France ou en Italie. « La boucle est bouclée », indique même un dernier « résumé » du site, qui souligne les nouvelles infections de Chine venant de l’étranger.
Trouver le meilleur jeu de paramètres
Ce n’est pas tout. D’autres outils mathématiques vont définitivement consacrer le rôle de la phylodynamique dans l’étude des pandémies. Si, en plus de l’horloge moléculaire et de la « généalogie », on ajoute la dynamique de l’épidémie sur le terrain, en prenant en compte le nombre de contacts, les durées d’incubation, de contamination, etc., les spécialistes peuvent alors tirer des soubresauts génétiques des informations que seuls les épidémiologistes détenaient jusqu’à présent. A savoir, en combien de temps le nombre de malades double-t-il ? Combien une personne en infecte-t-elle d’autres ? Voire, combien de personnes sont-elles malades ?
En effet, tous ces points ont une influence sur le virus lui-même. Une phase de croissance exponentielle d’une épidémie ne générera pas le même arbre généalogique ou phylogénétique qu’une maladie endémique. L’arbre du virus de la grippe, par exemple, est fort différent de celui du coronavirus. « Sur la grippe, on a des buissons et une forte compétition qui fait que, chaque année, une souche l’emporte sur les autres. Avec le SARS-CoV-2, nous ne voyons pas encore les effets potentiels de cette compétition, et les différents sous-groupes n’en sont peut être pas vraiment », décrit Olivier Gascuel, directeur de recherche au CNRS et à l’Institut Pasteur et membre de l’Académie des sciences.
Un algorithme cherche donc le meilleur jeu de paramètres, celui qui colle le mieux aux données génétiques. Si, pour le calcul des arbres, les logiciels PhyML, IQ-TREE ou RaxML se taillent la part du lion dans les analyses, cette seconde étape est dominée par Beast et Beast 2. L’équipe de Tanja Stadler, qui contribue au développement permanent de ces « bêtes » (beasts, en anglais), a très vite produit ses estimations des paramètres-clés de l’épidémie. Le 6 mars, par exemple, sur 128 génomes, ses estimations escomptent un taux de reproduction – c’est-à-dire le nombre de personnes infectées en moyenne par une première – compris entre 2 et 3,5, une valeur cohérente avec ce que dit l’épidémiologie classique. Elle estime aussi que, en Chine, au moment où 570 cas ont été officiellement recensés, le nombre réel était compris entre 2 000 et 30 000 cas.
Barres d’erreurs
Samuel Alizon s’est aussi livré à l’exercice pour la France. Selon ses calculs, l’ancêtre commun à la majorité des séquences françaises aurait émergé entre la mi-janvier et la mi-février. Le temps de doublement de l’épidémie serait passé de 2,5 jours au début de l’épidémie à 5 jours, si l’on prend en compte les malades plus récents. Des chiffres conformes à ceux tirés des courbes d’évolution du nombre de cas. Le taux de reproduction a lui aussi varié, ayant été divisé par deux entre la période du 21 février au 11 mars et celle du 19 mars au 22 mars, après le confinement. Evidemment, comme le rappelle le chercheur, ces résultats sont à prendre avec précaution, car le nombre de séquences est finalement petit, que ces séquences ne sont pas forcément représentatives et que les estimations ont des barres d’erreurs.
« Dans les années 1990, je me souviens que les gens ne voyaient pas la portée de ces techniques ni l’intérêt profond des reconstructions évolutives pour comprendre la biologie d’aujourd’hui. Pour eux, la phylogénie évoquait le vieux muséum d’histoire naturelle et l’époque de Darwin. Il faut dire aussi qu’il y avait peu de génomes séquencés», rappelle Olivier Gascuel, pionnier en France du domaine et coauteur du logiciel PhyML.
« Travailler sur des données en temps réel, c’est très motivant. Tout comme contribuer à dissiper le brouillard sur cette épidémie », souligne Jérémie Sciré, doctorant dans l’équipe de Tanja Stadler, qui participe au groupe de travail suisse sur le Covid-19. L’heure est aussi au partage des informations, comme le montrent les logiciels dont les codes sont ouverts, la profusion des séquences ou encore le forum « Virological.org », sur lequel les premières séquences ont été annoncées.
Conséquence : le domaine fait face à une crise de croissance. « Le principal défi est le passage à l’échelle. Des outils comme Beast ne peuvent pas traiter plus de 1 000 génomes ! », indique Olivier Gascuel, qui développe des techniques pour améliorer les algorithmes actuels et répondre à l’inflation. Même voie suivie par l’équipe de Tanja Stadler. Celle-ci veut aussi approfondir les liens nouveaux entre l’épidémiologie de terrain, qui construit des arbres de transmission, et la génétique, avec ses arbres généalogiques. « Nous pourrions détecter d’éventuels différentiels de transmission selon les sexes, ou retracer la propagation du virus entre différentes régions ou villes », aime à croire Samuel Alizon, dont le projet déposé sur ce sujet n’a été retenu qu’en liste complémentaire par l’Agence nationale de la recherche lors de son dernier appel d’offres. L’arbre de la phylodynamique doit encore trouver sa place dans la jungle de la recherche…

Aucun commentaire:
Enregistrer un commentaire