
Par Florence Rosier Publié le 9 mai 2022

RÉCIT Depuis le 31 mars, l’humanité dispose enfin d’un atlas presque complet de son génome – à l’exception du chromosome Y. Retour sur l’aventure d’exception que cette exploration a représentée. Un « pas de géant » non dépourvu de promesses abusives.
Le 16 février 2001, les revues Science et Nature annonçaient un exploit. L’intelligence humaine, enfin, était parvenue à lire le code qui programme son existence : le grand livre du génome humain. Autrement dit, le mode d’emploi de la construction d’un être humain. Un succès obtenu au terme de plus de dix années d’efforts, mobilisant une centaine de chercheurs et de techniciens à travers le monde.
Ce jour-là, la saga du projet Génome humain (HGP, pour Human Genome Project) entrait dans la légende. Mais l’annonce, en réalité, était survendue : cette édition originale restait un brouillon, truffé d’erreurs et parsemé de blancs. Des pages et des chapitres entiers faisaient défaut ! On mettra vingt ans, cependant, à comprendre à quel point elle était survendue, le temps de venir à bout de ce déchiffrage.
Au total, trente-huit rééditions de ce grand livre seront publiées au fil des années. Les fautes seront corrigées, les trous colmatés. Ainsi, le 31 mars, les chercheurs expliquaient, toujours dans Science, avoir lu les derniers 8 % de notre génome, jusque-là indéchiffrables. Soit, en taille, l’équivalent d’un petit chromosome. Fin de la saga ? Pas tout à fait, puisqu’il manque encore le chromosome Y, qui concerne la moitié – mâle – de l’humanité.
Pourquoi aura-t-il fallu tant de temps pour – presque – terminer cette lecture ? Quand le projet Génome humain a été lancé, fin 1998, l’entreprise relevait du défi. Notre génome est une encyclopédie en 46 volumes qui correspondent à nos vingt-trois paires de chromosomes. Son message est gravé sur un ruban de près de 2 mètres : notre ADN, enroulé et compacté dans le noyau de chacune de nos cellules. Il est écrit avec seulement quatre lettres différentes : les quatre bases de l’ADN, qui constituent l’alphabet biochimique des génomes, mais ce texte totalise trois milliards de lettres ! C’est donc cette interminable succession qu’il fallait déchiffrer, et dans le bon ordre. Une tâche fastidieuse quand le sens du message vous échappe. D’un côté, une chimie monotone ; de l’autre, une personne dans toute sa complexité. Vertigineux contraste…
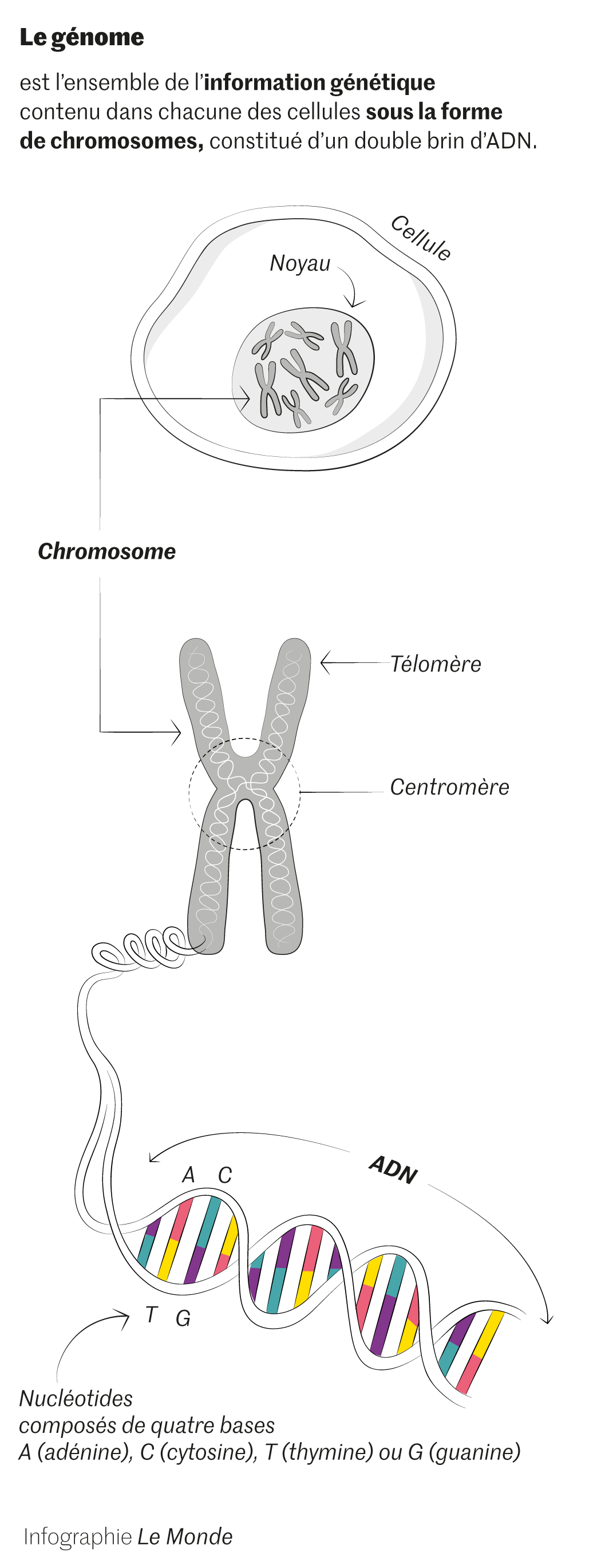
Saluée dans le monde entier, la prouesse de 2001 méritait cet hommage. Pour le symbole et pour le travail colossal accompli, en ce Moyen Age des techniques de lecture du génome. L’être humain, par cette plongée introspective en lui-même, a pu croire qu’il touchait là le sens de sa propre nature. Il a pu croire que la connaissance de son génome lui livrerait les clés de ses aptitudes et de leurs défaillances, les secrets de ses maladies et de leur guérison. Qu’elle augurerait l’avènement d’une nouvelle ère biomédicale, où la génomique toute-puissante permettrait de peaufiner une médecine de précision : des traitements personnalisés selon les fautes d’orthographe détectées dans le livre de chaque patient. Il a pu croire qu’elle lui offrirait la possibilité de corriger ses défauts et de reculer ses limites. Bref, « d’augmenter » l’espèce humaine. La machine à fantasmes était lancée.
Un pas de géant
« Pour les spécialistes de génétique médicale, la lecture de notre génome, dès 2001, a représenté un pas de géant », témoigne Stanislas Lyonnet, directeur de l’institut des maladies génétiques Imagine du campus Necker (AP-HP, Inserm, université Paris Cité). Là où il fallait, dans les années 1990, trois à dix ans d’efforts internationaux pour débusquer le gène responsable d’une maladie comme la mucoviscidose ou la myopathie de Duchenne, il suffit maintenant de lire le génome d’un patient et de le comparer au génome humain dit « de référence » – celui séquencé en 2001 ou, mieux, celui des rééditions ultérieures. « En un clic, nous pouvons localiser la région d’intérêt, identifier la protéine en jeu… »
Mais la promesse était un peu trop belle. « Si tromperie il y a eu, c’est surtout dans la survente de l’immédiateté des retombées médicales », poursuit Stanislas Lyonnet. Tests génétiques des maladies, thérapies géniques à tout-va, dépistage systématique des populations, préventions ciblées : « Pour les chercheurs qui ont réalisé le séquençage de 2001, le problème des maladies allait être réglé. Mais nous supposions bien que ce ne serait pas aussi évident », ajoute le généticien.
Pourtant, nombreux sont ceux à avoir été séduits. « Après mes études de médecine, en 1994, je me suis lancée à corps perdu dans la génétique en me disant : “C’est l’avenir.” On était dans un mouvement de folie », racontait au Temps Ariane Giacobino, professeure de génétique aux Hôpitaux universitaires de Genève, à l’occasion des 20 ans de la percée de 2001. « On a vu se dérouler la lecture du génome de l’homme, de la vache, du cochon, de la truffe du Périgord… C’était assez fascinant. » « Aujourd’hui, ajoute-t-elle, on est un peu revenu de cette génétique toute-puissante. La lecture des génomes a surtout révélé la béance de tout ce que nous ne parvenons pas à comprendre. » Le déterminisme de maladies chroniques aussi fréquentes que le diabète, les affections psychiatriques ou cardiovasculaires, par exemple, nous échappe encore.
C’est que les généticiens ont buté sur un écueil inattendu : la diversité des génomes, au sein des populations humaines. « Entre vous et moi, le génome diffère de 0,1 % en moyenne, note Stanislas Lyonnet. A première vue, c’est peu. Mais cela correspond à trois millions de lettres différentes ! » C’est ce qu’a révélé le consortium 1 000 génomes en 2010. Et c’est ce qui a compliqué l’interprétation des données du génome de chaque patient.
Et puis, ce fameux 16 février 2001 a aussi marqué le début d’une polémique, toujours pas close, entre les deux équipes qui annonçaient le même exploit. Nature publiait le travail d’un consortium international de chercheurs académiques, le fameux HGP, coordonné par le généticien américain Francis Collins. La revue Science, elle, annonçait le résultat d’un franc-tireur, le biotechnologiste Craig Venter, sorte d’Elon Musk du génome, qui avait mobilisé sa société, Celera Genomics. Alors que le HGP avait été lancé en 1989, Craig Venter n’avait commencé à relever le défi qu’en 1998.
Au bout du compte, il a fallu onze ans au projet public, contre trois au projet privé, épaulé par une robotisation massive, pour franchir la ligne d’arrivée. Ensemble, donc. Mais Celera avouera avoir utilisé, en sus de ses données, celles publiées en ligne par le consortium international à mesure de ses avancées. Un pillage, s’indignera-t-on. Public contre privé, démarche altruiste ou mercantile : cette opposition sera ravivée six ans plus tard. Dès 2007, des sociétés comme 23AndMe ou Ancestry DNA, en effet, se sont mises à commercialiser en ligne des analyses du génome, pour une centaine de dollars. Leurs clients : des particuliers désireux de connaître leurs origines généalogiques ou – ce qui suscitera de vifs débats sur la fiabilité des résultats – leurs risques de maladies…
La question du génome de référence
La percée de 2001 a aussi soulevé une question : qu’est-ce qu’un « génome de référence » ? « C’est juste une carte utile pour positionner les éléments fonctionnels de l’ADN que les chercheurs étudient », explique Laurent Duret, spécialiste de biologie évolutive au CNRS à l’université de Lyon. C’est « le GPS du génome », résume Stanislas Lyonnet.
Dans le projet académique, ce génome de référence était « un génome mosaïque de cinq individus », précise Laurent Duret. Les chercheurs avaient prélevé l’ADN de cinq personnes de différentes ethnies, puis découpé le génome de chacun d’eux en fragments, ils les avaient ensuite lus puis réassemblés. Toutes les rééditions suivantes seront établies à partir de ces cinq mêmes individus. Quant au génome publié par Celera, censé inclure plusieurs individus, il était essentiellement constitué du génome… de Craig Venter.
Le génome de référence permettra aussi de recenser le nombre de nos gènes dits « codants », ceux qui délivrent les instructions nécessaires à la fabrication des protéines de tous nos tissus et organes. Ces protéines jouent un double rôle clé. Ce sont des briques élémentaires qui participent à la construction de nos cellules. Et ce sont aussi des rouages qui assurent leur bon fonctionnement, sous forme d’enzymes, de récepteurs… Eh bien, notre espèce compte près de 20 000 gènes codants (19 969, selon le dernier recensement), révélera le HGP.
L’exploration de notre génome a emprunté d’autres pistes. En 2003, un autre projet sera lancé, baptisé Encode (Encyclopedia of DNA Elements). Son but : identifier tous les éléments fonctionnels présents dans notre génome. Ses premiers résultats seront publiés dans Nature en 2012. Principal verdict : nos 20 000 gènes codants ne sont pas seuls à être noyés dans un océan d’ADN inutile, nommé « ADN poubelle ».
Ces 20 000 gènes, en effet, ne représentent que 2 % de notre ADN. Pourquoi donc notre génome est-il à ce point hypertrophié ? En réalité, il comporte plus de régions fonctionnelles qu’on ne le pensait, révélera Encode. « Il contient aussi 43 500 gènes “non codants”, soit 4 % du génome. Ces gènes-là commandent non pas la production de protéines, mais celle de molécules d’ARN essentielles au bon fonctionnement de nos cellules : ARN de transfert, ARN ribosomaux, micro-ARN… », explique Stanislas Lyonnet. Plus encore, notre génome comporte entre 10 % et 15 % de régions très conservées d’une espèce à l’autre, sur le plan évolutif. C’est le signe de l’importance de leur mission : elles contrôlent l’activité de nos gènes, pour tantôt les « allumer », tantôt les « éteindre ». Pour autant, « il reste encore de 85 % à 90 % de notre génome sans aucun signe de fonctionnalité. Le projet Encode n’a donc pas sonné le glas de l’ADN poubelle, comme on a pu le croire », relève Laurent Duret.
Il est temps maintenant de parler un peu technique. Dans les années 1990, la lecture de l’ADN était moyenâgeuse. Selon une bonne vieille technique mise au point par Fred Sanger, un biochimiste anglais qui recevra deux fois le Nobel, l’ADN à lire était découpé en fragments. Puis on en fabriquait des brins miroirs, que l’on repérait par l’intégration de lettres de l’ADN marquées au phosphore radioactif. Enfin, on faisait migrer le tout sur un gel. La procédure, très chronophage, consommait énormément de gel et d’ADN.
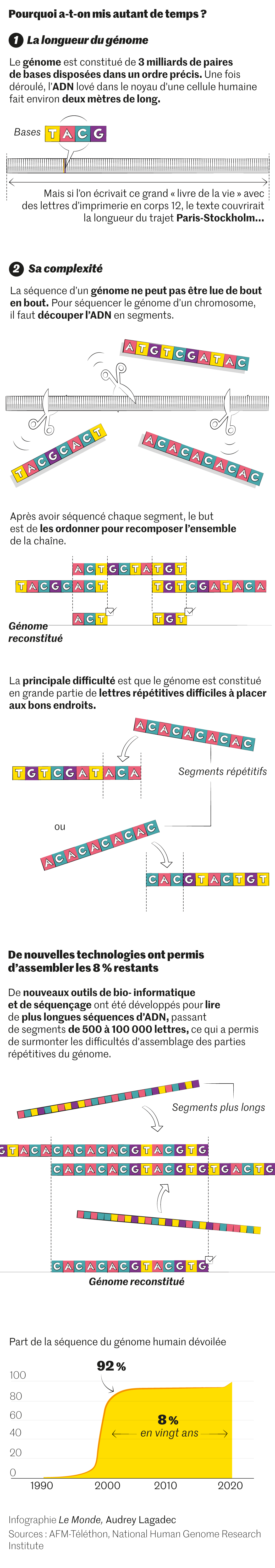
Au milieu des années 2000, une technologie de rupture est apparue. Une comparaison en donne la mesure. Pour dresser la première séquence complète du génome humain, il aura donc fallu onze ans d’efforts internationaux, pour un coût de 1 milliard de dollars (950 millions d’euros). Aujourd’hui, le génome d’un individu peut être séquencé en quelques jours, de façon fiable, pour quelques centaines de dollars – en réalité, on séquence seulement les régions codantes de son génome.
Ce vertigineux progrès est dû au séquençage de nouvelle génération (NGS, pour Next Generation Sequencing). On le doit notamment à un Français, Pascal Mayer, et à deux Britanniques, Shankar Balasubramanian et David Klenerman. Tous trois ont, en 2021, reçu pour cette avancée le Breakthrough Prize in Life Sciences, qui passe pour être l’antichambre du Nobel.
« Pour progresser, il [a fallu] réunir les compétences de biochimistes, de biologistes moléculaires, de biophysiciens, d’experts en optique, en microfluidique, en traitement du signal et en informatique », Pascal Mayer, biophysicien
« Pour progresser, il [a fallu] réunir les compétences de biochimistes, de biologistes moléculaires, de biophysiciens, d’experts en optique, en microfluidique, en traitement du signal et en informatique », confiait au Monde Pascal Mayer, en octobre 2021. Et ce fut un festival d’innovations. Jugez-en : le génome à lire est d’abord découpé en tout petits fragments d’ADN, qui sont ensuite fixés sur une lame (à raison de 10 000 à 100 000 fragments par millimètre carré). Puis chaque fragment est lu en parallèle. Comme si, au lieu de lire un livre de la première à la dernière page, on lisait chacune de ses pages en parallèle. La suite fait appel à l’utilisation de lettres de l’ADN fluorescentes et à la spectrométrie de fluorescence. Enfin, des algorithmes spécifiques permettent de reconstituer le puzzle : comme ces fragments se chevauchent, ils peuvent être alignés dans le bon ordre.
Cette géniale innovation sera d’une redoutable efficacité pour lire… 92 % de notre génome. Mais elle sera mise en échec par quelques pages, enfouies dans des recoins du grimoire. Au total, 8 % de notre génome restaient illisibles. Ces pages sont ultra-compactes – elles appartiennent à des régions de l’ADN ultra-condensées et pauvres en gènes codants. Et, surtout, elles sont couvertes de mots ultra-répétés. Une fois leur ADN découpé en petits fragments, ceux-ci se ressemblent tous : leur réassemblage devient un casse-tête.
Il faut dire que notre génome a une étrange propriété dont nous n’avons pas encore parlé : il comporte une importante proportion – environ 45 % – de séquences d’ADN qui se répètent (comme un même mot réitéré). Parmi ces séquences, certaines sont ultra-répétées (des dizaines de milliers de fois) : ce sont les fameux 8 % restés illisibles.
Il a donc fallu encore ruser. Ou, plus exactement, profiter de l’arrivée, au milieu des années 2010, d’une nouvelle méthode de lecture du génome. Avec elle, l’ADN n’est plus découpé en myriades de petits fragments (comptant chacun environ 100 lettres), mais en fragments de mille à dix mille fois plus longs. Le réassemblage des pièces du puzzle, aidé d’algorithmes ultra-performants, est alors bien plus facile.
Grâce à cela, les « poubelles » de notre génome ont pu être fouillées. Leur ADN ultra-répété, enfin, a été lu et ordonné par un autre consortium, le Telomere-to-Telomere (T2T). Créé en 2018, il associe trente-trois institutions et universités, pour l’essentiel américaines. Son but : séquencer nos chromosomes d’un bout à l’autre – télomère étant le nom de l’extrémité des chromosomes.
ADN d’origine virale
Leur lecture permet d’explorer leurs mystérieuses fonctions. Ce sont des « cimetières d’éléphants », nous expliquait Stanislas Lyonnet, en mars. Sauf que ces éléphants sont souvent des virus, disparus depuis des lustres. Jadis, ils ont infecté nos lointains ancêtres. Et ils en ont profité – un abus de langage, car l’évolution est opportuniste, mais pas finaliste – pour intégrer une partie de leur matériel génétique dans le génome des personnes infectées. « Quand ce processus a touché les cellules germinales, cet ADN d’origine virale, intégré dans le génome humain, a été transmis aux descendants des personnes infectées », explique Laurent Duret. Sans, la plupart du temps, bénéficier à leurs hôtes : bel exemple d’ADN égoïste, dont le seul but est de se reproduire.
« Environ 90 % des séquences ultra-répétées de notre génome se trouvent dans les centromères », ajoute le chercheur. Les centromères ? Ce sont ces régions d’ADN situées au centre du X que forment nos chromosomes. Enfin, presque au centre pour la majorité d’entre eux – et pas du tout au centre pour les numéros 13, 14, 15, 21 et 22. Les centromères jouent un rôle crucial lors de la division de nos cellules. « Ce sont eux qui assurent la répartition équilibrée des chromosomes dans chaque cellule fille », explique Daniele Fachinetti, directeur d’une équipe CNRS à l’Institut Curie (Paris).
C’est sur les centromères, en effet, que s’ancre la machinerie qui sépare les paires de chromosomes, lors de toute division cellulaire. Dans ce processus, un complexe d’une centaine de protéines, le kinétochore, joue un rôle majeur. Il s’arrime solidement au centromère afin que d’autres rouages, à l’intérieur de la cellule, puissent attraper, puis séparer les paires de chromosomes. Quand ce processus se passe bien, chaque cellule fille hérite du nombre correct de chromosomes.
Mais « quand ce processus se passe mal, on se retrouve avec des chromosomes mal séparés, ce qui entraîne toutes sortes de problèmes », explique Nicolas Altemose, chercheur postdoctoral à l’université de Berkeley (Californie), coauteur de quatre des articles dans Science. « Si cela se produit lors de la méiose [la formation des spermatozoïdes et des ovules], cela peut provoquer des anomalies chromosomiques, entraînant des fausses couches spontanées ou des maladies congénitales. Si cela se produit dans les cellules somatiques [les cellules de tous nos organes, à l’exception des cellules sexuelles], cela peut entraîner un cancer. » C’est notamment le cas lorsque certains gènes du centromère sont surexprimés. Résultat, les cellules cancéreuses se divisent de façon anarchique.
« Dans environ neuf tumeurs solides sur dix, on retrouve une anomalie du nombre de chromosomes. Et dans six cancers du sang sur dix », révèle Daniele Fachinetti. En sus des cancers, d’autres maladies sont liées à des anomalies touchant des séquences répétées : des maladies héréditaires (hémophilies, épilepsies…) ou des désordres génomiques.
Autre découverte du T2T : dans les 8 % nouvellement séquencés, les chercheurs ont débusqué environ 2 000 nouveaux gènes codants (ils appartenaient à des familles déjà connues). La plupart sont désactivés, mais 115 peuvent être encore actifs. Les chercheurs ont aussi trouvé deux millions de variants supplémentaires, dont 622 dans des gènes importants au plan médical. « On ne sait pas trop ce que les centromères apportent à l’expression des caractères propres à chaque individu, confiait Philippe Froguel, professeur de génétique à l’Imperial College de Londres, en mars. L’étude de leurs variants va aider à répondre. »
Alors, arrivons-nous au terme de la longue saga du déchiffrage de notre génome ? C’est oublier qu’en science chaque découverte suscite une nouvelle interrogation. Prochaine étape : lire l’intégralité de notre épigénome, cet assemblage de marques chimiques (méthyles, protéines, ARN…) accolées à notre ADN.
Leur mission est capitale : elles assurent durablement, dans chacune de nos cellules, l’activation ou l’inhibition d’un panel de nos gènes. Or, ce panel diffère selon les types de cellules : c’est ce qui explique pourquoi une cellule de notre cerveau diffère d’une cellule de notre intestin, alors qu’elles portent toutes deux le même génome. Voilà pourquoi la lecture de notre épigénome devra être répétée pour chaque type de cellules. Vaste programme.
Aucun commentaire:
Enregistrer un commentaire